
A computational pipeline for cross-species cancer analyses
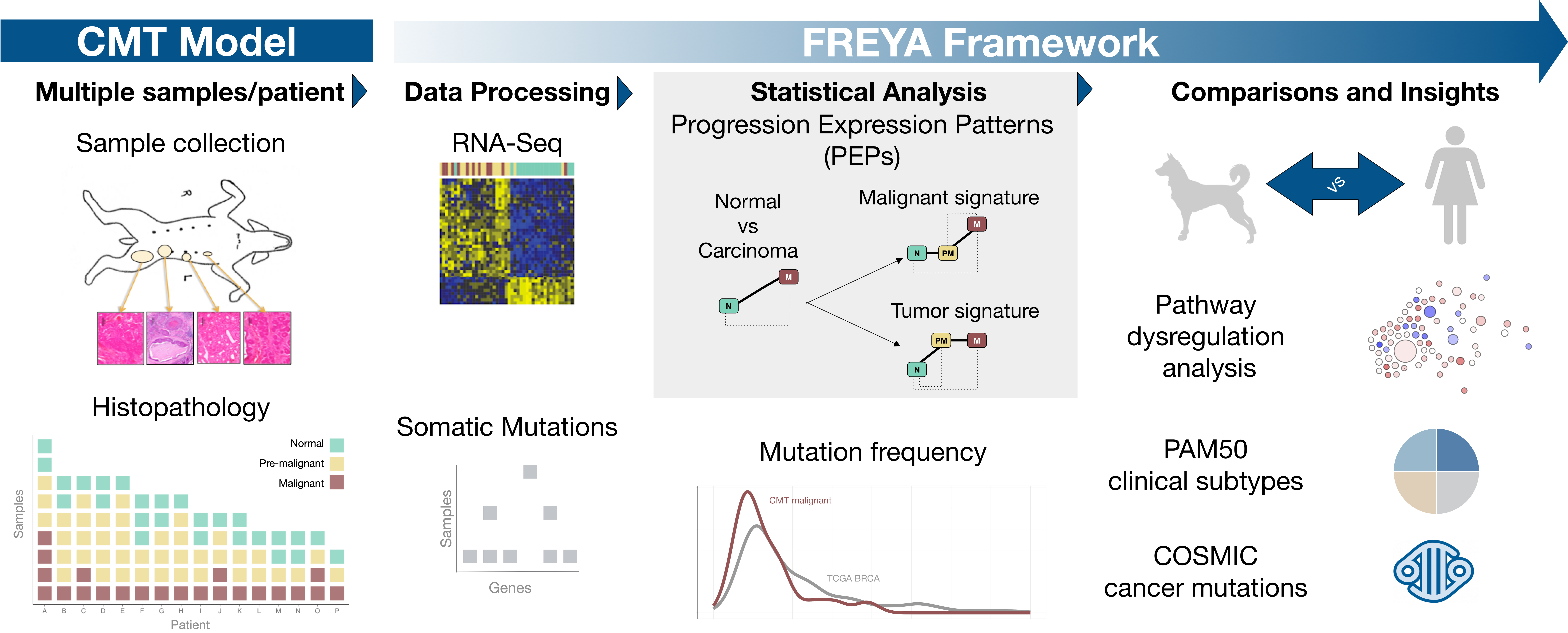
The FREYA analytic framework calculates genetic diversity within a cancer cohort, extracts progression-related patterns of expression, calls functional variants, identifies intrinsic human molecular tumor subtypes, and compares them to human breast cancer biology. It helps streamline future canine mammary tumor (or other tissue/species) analyses and provides a comprehensive suite of tools that encompass conventional human analyses and new dog-centric approaches, seamlessly integrating the two.
FREYA is split into two major components: data preparation (DataPrep) and analysis (DataAnalysis). DataPrep prepares sequencing data for analysis. DataAnalysis runs statistical analyses from the manuscript. Both DataPrep and DataAnalysis are designed to run with user-provided data. The pipeline requires that each patient (e.g. each dog) has at least 1 sample of each histology: normal, benign, and malignant.
 Install FREYA
Install FREYA ( DataPrep )
DataPrep is the first sub-pipeline of FREYA. It will prepare processed expression and mutation call data, using the raw data downloaded from your sequencer. DataPrep runtime is highly dependent on the machine used. For example, the dataset used in the CMT manuscript takes approximately 1 month to process using 1 cpu but can be run in less than 24 hours using the DisBatch setup.
This pipeline has several dependencies, which are, in effect, described by the Dockerfile. The central script is "cmwf_csv.sh." Please see our GitHub documentation for more information. Mutations are called using GATK Best Practices workflow for SNP and indel calling on RNAseq data.
Install FREYA
( DataAnalysis )
This second sub-pipeline depends on processed genomic and phenotype data (created either by yourself or by using FREYA DataPrep). If you used DataPrep, be sure to first run the prep_data.R script to convert the gene names etc. If you are providing pre-processed data you may be able to skip this step (see below).
FREYA DataAnalysis can be run from your browser using either Docker and the Jupiter notebooks, or on the command line by using the provided Makefile. Update the variables in the Jupyter notebook, following the order in Index.ipynb, or in the Makefile, to reflect your data files. We provide some example simulated data in the synthetic_data folder. If you want to view or run the pipeline without using your own data, you can do so by clicking Launch Binder (also in the Github repository).
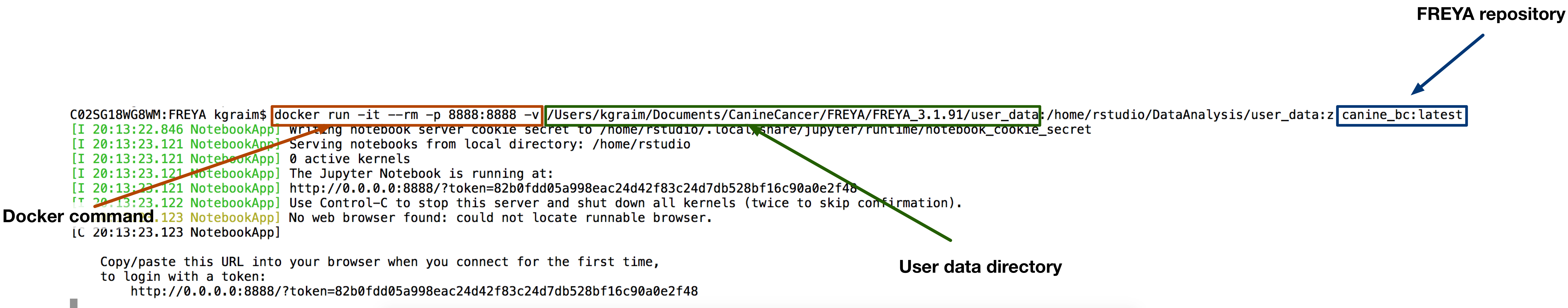
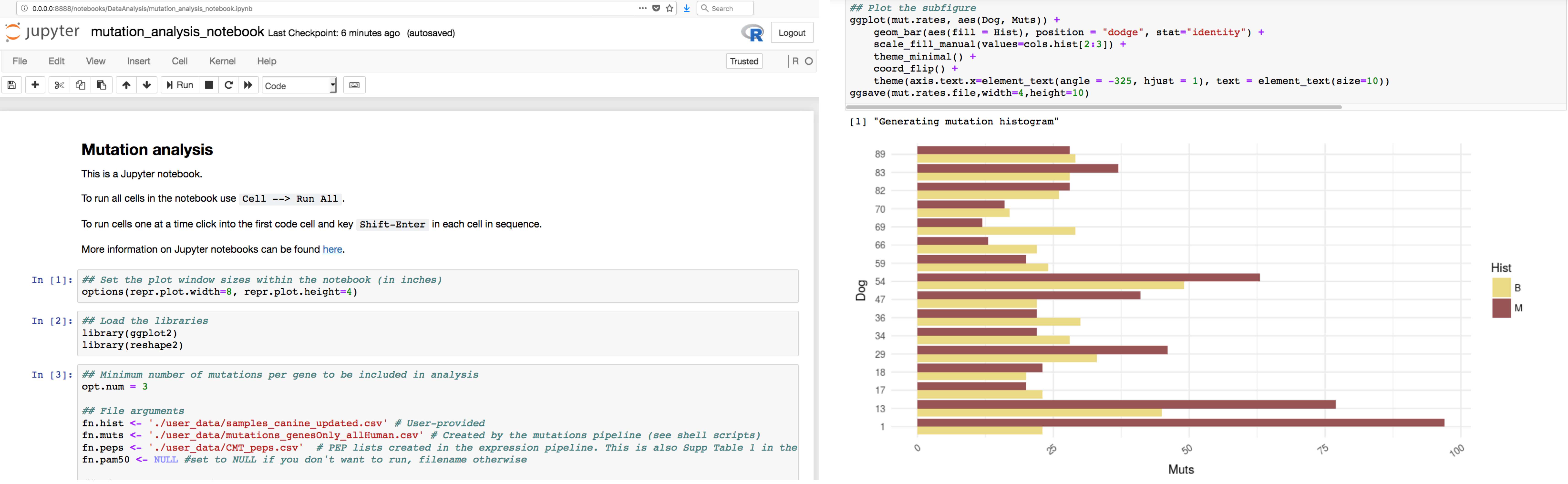 Install FREYA
Install FREYA